What are pigmented fundus lesions?
During an eye examination, optometrists look at the inside of the eye to make sure it is healthy. Sometimes they spot changes that might need monitoring or more tests.
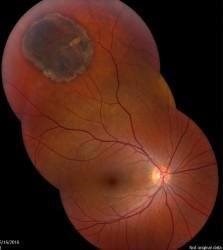
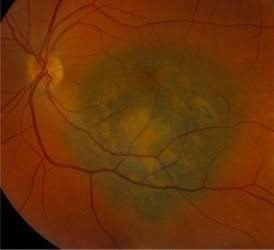
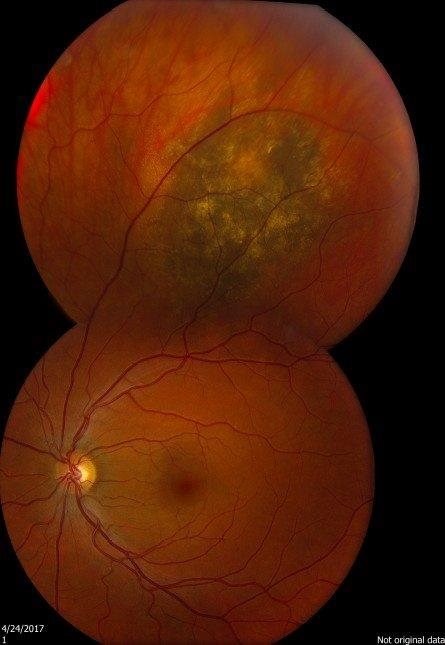
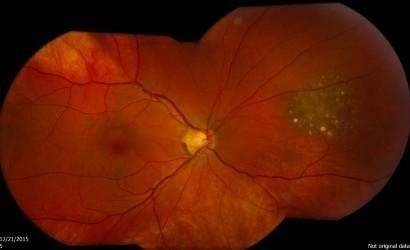
One change that is quite often seen is called a choroidal naevus. This is like a mole on the skin and is found in a part of the eyeball called the choroid. The choroid is the layer of nourishing and supportive tissue at the back of the eye between the retina (the part of the eye that receives light and sends images to the brain) and the outer coat of the eye. A choroidal naevus cannot be seen from the outside of the eye and usually causes no symptoms. Without professional eye examinations, people may never know they have one. If a choroidal naevus is found, the optometrist will check it at each regular eye examination.
If your choroidal naevus does not look normal, it is called an atypical choroidal naevus. This will be monitored by your ophthalmologist or optometrist through regular eye examinations as there is a small chance it may change into a cancer. They will take photographs of the inside of your eye so that they can see if your choroidal naevus changes or grows over time.
Some people are born with a harmless blemish inside the eye called Congenital Hypertrophy of the Retinal Pigment Epithelium (CHRPE). This can be confused with a choroidal naevus, but will not need to be regularly monitored.
Sometimes the optometrist will see a change inside the eye that might be a cancer called choroidal melanoma. Choroidal melanoma is very rare.
How are pigmented fundus lesions managed?
If an optometrist thinks someone might have developed choroidal melanoma, they will refer that person to an ophthalmologist (a specialist eye doctor) within two weeks. The ophthalmologist will examine the eye and carry out some tests to see if there is a cancer.
A person diagnosed with choroidal melanoma will be offered treatment to reduce the risk of it spreading to other parts of the body and to save as much of the vision as possible. The treatment will depend on the type of cancer that is found and how advanced it is. More information about choroidal melanoma and different treatments can be found on Macmillan Cancer Support’s website.